7.1.3. Mikado serialise¶
Mikado integrates data from multiple sources to select the best transcripts. During this step, these sources are brought together inside a common database, simplifying the process of retrieving them at runtime. Currently, Mikado integrates three or more different types of data:
- reliable junctions, in BED12 format
- ORF data, currently identified using TransDecoder or Prodigal. Any program capable of generating it in BED12 or GFF3 format is suitable.
- BLASTX data (from [Blastplus] or [Diamond])
- Miscellaneous numeric scores can also be given as input to Mikado in a tabular file
After serialisation in the database, these sources will be available to use for any subsequent Mikado pick run. Having the data present in the database allows to run Mikado with multiple configurations and little overhead in terms of pre-loading data; this feature is taken directly advantage of in The Daijin pipeline for driving Mikado, where it is possible to run Mikado using multiple modes.
Mikado serialise can use three different SQL databases as backends - SQLite, MySQL and PostgreSQL - thanks to SQLAlchemy. This step, together with the creation of the TransDecoder and BLAST data, is the most time consuming of the pipeline. In particular, although Mikado serialise will try to analyse the XML data in a parallelised fashion if so instructed, the insertion of the data in the database will still happen in a single thread and will therefore be of limited speed. If using SQLite as database (the default option), it is possible to decrease the runtime by modifying the “max_objects” parameters, at the cost however of increased RAM usage.
Important
The schema of Mikado databases changed with version 1.3. Any database created prior to this version should be regenerated, otherwise Mikado pick will fail.
7.1.3.1. BLAST files¶
Mikado serialise is capable of using homology results derived from either BLAST+ [Blastplus] or DIAMOND. Further, it is capable of analysing results provided in multiple formats:
- Tabular BLAST file. This is now the preferred format. The file needs to have been created with the following field specification: - “qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore ppos btop”
- XML BLAST.
- ASN files, i.e. the BLAST+ archive format. These will be converted on-the-fly to tabular files with the appropriate keys.
- DAA files, i.e. the DIAMOND archive format. These will be converted on-the-fly to tabular files with the appropriate keys.
Warning
ASN files will need BLAST+ to be installed in the same environment/instance of Mikado, otherwise the on-the-fly conversion will fail. Likewise, DAA files require DIAMOND to be installed in the same environment/instance of Mikado.
All possible input files can be optionally compressed using GZIP or BZIP2; Mikado will handle the conversion. The only exception are DAA files, as these are already compressed by default.
Hint
The most expensive operation in a “Mikado serialise” run is by far the serialisation of BLAST XML files. Using tabular files significantly speeds up the process, as they are much smaller and faster to read and parse. In case XML files are used, splitting the input files in multiple chunks, and analysing them separately, allows for better parallelisation. If a single monolythic XML/ASN file is produced, by contrast, Mikado will be quite slow.
7.1.3.2. Transdecoder ORFs¶
When Mikado analyses ORFs produced by TransDecoder, Prodigal or equivalent program, it performs additionally the following checks:
- Check the congruence between the length of the transcript in the BED12 file and that found in the FASTA file
- Check that the ORF does not contain internal stop codons
- Check that the CDS length is valid, ie a multiple of 3, if the ORF is complete
- Optionally, if the ORF is open on the 5’ side, Mikado can try to find an internal start codon. See this section for details.
Mikado can accept the data in GFF3 format (the standard for Prodigal) or BED12 (preferred for TransDecoder).
7.1.3.3. Reliable junctions¶
Some aligners (e.g. STAR) are capable of returning a list of splice junctions, inferred by the read alignments, that they repute to be extremely reliable. Likewise, our tool Portcullis is capable of analysing one or multiple BAM alignment files and return a list of junctions that are well-supported by the data (e.g. high coverage of diverse reads around the junction, with few or no mismatches) and therefore to be considered more trustworthy from the rest. Mikado actively makes use of this splice junction data to score and select transcripts.
We require the data to be provided in BED12 format (see sidebar for Mikado-specific details). Portcullis provides utilities for converting junction data and/or merge multiple datasets into a single file, through its junctools command line utility.
Additionally, Mikado is capable of interpreting BED12 files of manually curated genes as a source of reliable junctions. Please see the sidebar for details.
7.1.3.4. Additional scores¶
Aside from the above datasets, Mikado can further integrate scores from different sources, such as structural coherence with known annotations from other species, coding-potential calculations, or expression data. To do so, Mikado needs a tab-delimited file with all transcript IDs present in the first column, which should be marked as tid. As an example of a valid file, with two columns (tpm ie expression data, and CPC ie coding potential):
| tid tpm CPC |
| at_AT5G66600.2 24260.8 1 |
| at_AT5G66600.3 121.857 1 |
| at_AT5G66600.4 0 1 |
| at_AT5G66600.1 4775.2 1 |
| cuff_cufflinks_star_at.23553.1 6358.42 .7 |
| cl_Chr5.6272 0 .3 |
| cl_Chr5.6271 0 .2 |
Our pipeline Minos, in development, creates and uses such tables to prioritise transcripts more effectively.
7.1.3.5. Usage¶
mikado serialise allows to override some of the parameters present in the configuration file through command
line options, eg the input files. Notwithstanding, in the interest of reproducibility we advise to configure everything
through the configuration file and supply it to Mikado prepare without further modifications.
Available parameters:
Parameters related to performance:
- start-method: one of fork, spawn, forkserver. It determines the multiprocessing start method. By default, Mikado will use the default for the system (fork on UNIX, spawn on Windows).
- procs: Number of processors to use.
- single-thread: flag. If set, Mikado will disable all multithreading.
- max_objects: Maximum number of objects to keep in memory before committing to the database. See this section of the configuration for details.
Basic input data and settings:
- output-dir: directory where the SQLite database and the log will be written to.
- transcripts: these are the input transcripts that are present on the GTF file considered by Mikado. Normally this should be the output of Mikado prepare.
- genome_fai: FAIDX file of the genome FASTA. If not given, serialise will derive it from the “reference: genome” field of the configuration.
- force: flag. If set, and the database is already present, it will be truncated rather than updated.
- json-conf: this is the configuration file created with Mikado configure.
- db: if the database is specified on the command line,
mikado serialisewill interpret it as a SQLite database. This will overwrite any setting present in the configuration file.
Parameters related to logging:
- log: log file. It defaults to
serialise.log. - log_level: verbosity of the logging. Please be advised that excessive verbosity can negatively impact the performance of the program - the debug mode is extremely verbose.
- log: log file. It defaults to
Parameters related to reliable junctions:
- junctions: a BED12 file of reliable junctions.
Parameters related to the treatment of ORF data:
- orfs: ORF BED12 / GFF3 files, separated by comma.
- max-regression: A percentage, expressed as a number between 0 and 1, which indicates how far can Mikado regress along the ORF to find a valid start codon. See the relative section in the configuration for details.
- no-start-adjustment: if selected, Mikado will not try to correct the start position in ORFs and will consider them as provided.
- codon-table: this parameter specifies the codon table to use for the project. Mikado by default uses the NCBI codon table 1 (standard with eukaryotes) with the modification that only ATG is considered as a valid start codon, as ORF predictions usually inflate the number of non-standard starts.
Parameters related to BLAST data:
- blast_targets: BLAST FASTA database.
- xml: BLAST files to parse. This can be either a comma-seprated list of valid files, or a comma-separated list of folders containing valid files. Please note that, notwithstanding the name of the flag, Mikado will also accept valid TSV files here (see above).
- max-target-seqs: maximum number of BLAST targets that can be loaded per sequence, for each BLAST alignment. Please note that if you align against multiple databases, this threshold will be applied once per file.
- Parameters related to additional scores:
- external-scores: a tab-delimited file of additional scores for the transcripts; one row per transcript.
Warning
It is advised to set this parameter to spawn even on UNIX. See the dedicated sidebar for details.
Usage:
$ mikado serialise --help
usage: Mikado serialise [-h] [--start-method {fork,spawn,forkserver}] [--orfs ORFS] [--transcripts TRANSCRIPTS] [-mr MAX_REGRESSION] [--codon-table CODON_TABLE] [-nsa] [--max-target-seqs MAX_TARGET_SEQS]
[-bt BLAST_TARGETS] [--xml XML] [-p PROCS] [--single-thread] [--genome_fai GENOME_FAI] [--genome GENOME] [--junctions JUNCTIONS] [--external-scores EXTERNAL_SCORES] [-mo MAX_OBJECTS]
[--no-force | --force] [--configuration CONFIGURATION] [-l [LOG]] [-od OUTPUT_DIR] [-lv {DEBUG,INFO,WARN,ERROR} | --verbose | --quiet | --blast-loading-debug]
[--seed SEED | --random-seed]
[db]
optional arguments:
-h, --help show this help message and exit
--start-method {fork,spawn,forkserver}
Multiprocessing start method.
--no-force Flag. If set, do not drop the contents of an existing Mikado DB before beginning the serialisation.
--force Flag. If set, an existing databse will be deleted (sqlite) or dropped (MySQL/PostGreSQL) before beginning the serialisation.
-od OUTPUT_DIR, --output-dir OUTPUT_DIR
Output directory. Default: current working directory
-lv {DEBUG,INFO,WARN,ERROR}, --log-level {DEBUG,INFO,WARN,ERROR}
Log level. Default: derived from the configuration; if absent, INFO
--verbose
--quiet
--blast-loading-debug
Flag. If set, Mikado will switch on the debug mode for the XML/TSV loading.
--seed SEED Random seed number. Default: 0.
--random-seed Generate a new random seed number (instead of the default of 0)
--orfs ORFS ORF BED file(s), separated by commas
--transcripts TRANSCRIPTS
Transcript FASTA file(s) used for ORF calling and BLAST queries, separated by commas. If multiple files are given, they must be in the same order of the ORF files. E.g. valid command
lines are: --transcript_fasta all_seqs1.fasta --orfs all_orfs.bed --transcript_fasta seq1.fasta,seq2.fasta --orfs orfs1.bed,orf2.bed --transcript_fasta all_seqs.fasta --orfs
orfs1.bed,orf2.bed These are invalid instead: # Inverted order --transcript_fasta seq1.fasta,seq2.fasta --orfs orfs2.bed,orf1.bed #Two transcript files, one ORF file
--transcript_fasta seq1.fasta,seq2.fasta --orfs all_orfs.bed
-mr MAX_REGRESSION, --max-regression MAX_REGRESSION
"Amount of sequence in the ORF (in %) to backtrack in order to find a valid START codon, if one is absent. Default: None
--codon-table CODON_TABLE
Codon table to use. Default: 0 (ie Standard, NCBI #1, but only ATG is considered a valid start codon.
-nsa, --no-start-adjustment
Disable the start adjustment algorithm. Useful when using e.g. TransDecoder vs 5+.
--max-target-seqs MAX_TARGET_SEQS
Maximum number of target sequences.
-bt BLAST_TARGETS, --blast-targets BLAST_TARGETS, --blast_targets BLAST_TARGETS
Target sequences
--xml XML, --tsv XML BLAST file(s) to parse. They can be provided in three ways: - a comma-separated list - as a base folder - using bash-like name expansion (*,?, etc.). In this case, you have to enclose
the filename pattern in double quotes. Multiple folders/file patterns can be given, separated by a comma. BLAST files must be either of two formats: - BLAST XML - BLAST tabular
format, with the following **custom** fields: qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore ppos btop
-p PROCS, --procs PROCS
Number of threads to use for analysing the BLAST files. This number should not be higher than the total number of XML files.
--single-thread Force serialise to run with a single thread, irrespective of other configuration options.
--genome_fai GENOME_FAI
--genome GENOME
--junctions JUNCTIONS
--external-scores EXTERNAL_SCORES
Tabular file containing external scores for the transcripts. Each column should have a distinct name, and transcripts have to be listed on the first column.
-mo MAX_OBJECTS, --max-objects MAX_OBJECTS
Maximum number of objects to cache in memory before committing to the database. Default: 100,000 i.e. approximately 450MB RAM usage for Drosophila.
--configuration CONFIGURATION, --json-conf CONFIGURATION
-l [LOG], --log [LOG]
Optional log file. Default: stderr
db Optional output database. Default: derived from configuration
7.1.3.6. Technical details¶
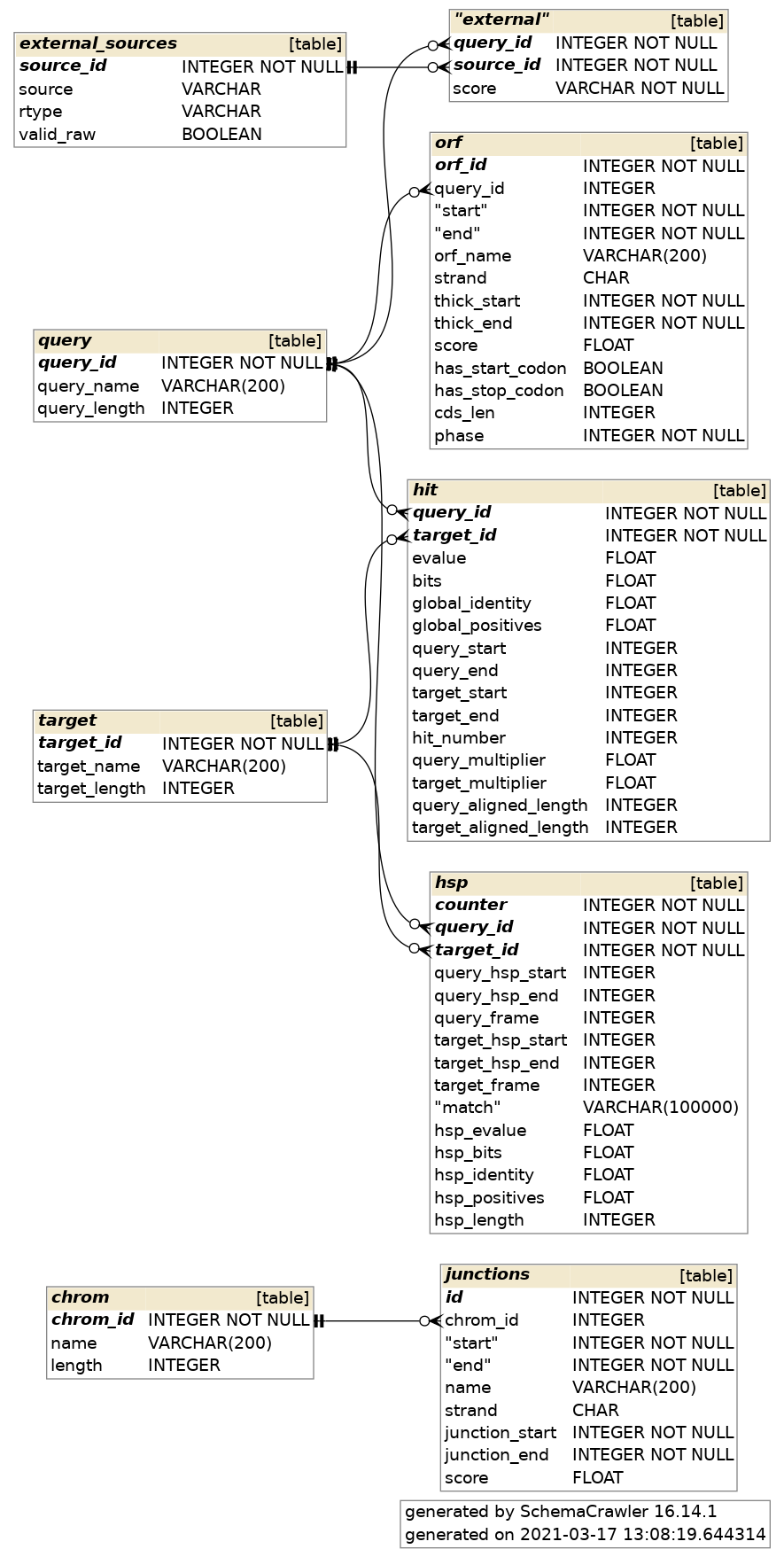
The schema of the database is quite simple, as it is composed only of 9 discrete tables in two groups. The first group, chrom and junctions, serialises the information pertaining to the reliable junctions - ie information which is not relative to the transcripts but rather to their genomic locations. The second group serialises the data regarding ORFs, BLAST files and external arbitrary data. The need of using a database is mainly driven by the need to avoid calculating all necessary information at runtime every time mikado pick is launched.
Database schema used by Mikado.