8. Mikado core algorithms¶
8.1. Picking transcripts: how to define loci and their members¶
The Mikado pick algorithm
Schematic representation of how Mikado unbundles a complex locus into two separate genes.
Transcripts are scored and selected according to user-defined rules, based on many different features of the transcripts themselves (cDNA length, CDS length, UTR fraction, number of reliable junctions, etc.; please see the dedicated section on scoring for details on the scoring algorithm).
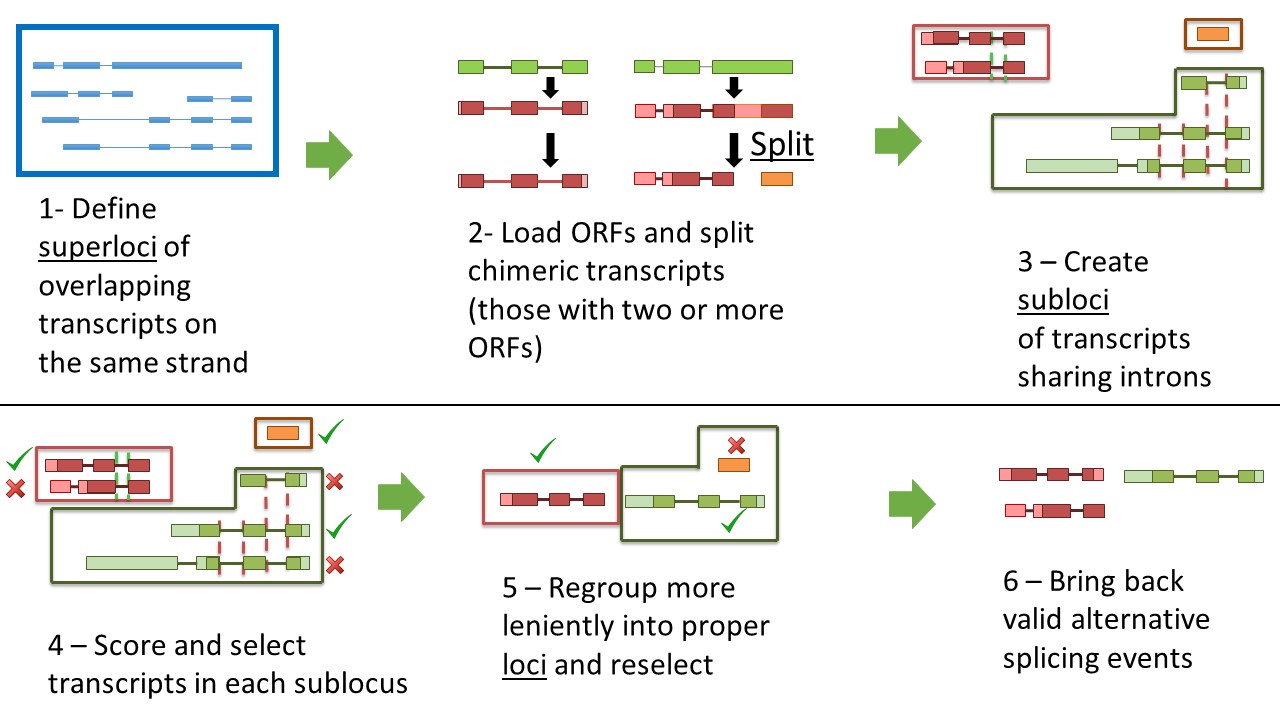
The detection and analysis of a locus proceeds as follows:
When the first transcript is detected, Mikado will create a superlocus - a container of transcripts sharing the same genomic location - and assign the transcript to it.
While traversing the genome, as long as any new transcript is within the maximum allowed flanking distance, it will be added to the superlocus.
When the last transcript is added, Mikado performs the following preliminary operations:
Integrate all the data from the database (including ORFs, reliable junctions in the region, and BLAST homology).
If a transcript is monoexonic, assign or reverse its strand if the ORF data supports the decision
If requested and the ORF data supports the operation, split chimeric transcripts - ie those that contain two or more non-overlapping ORFs on the same strand.
Split the superlocus into groups of transcripts that:
- share the same strand
- have at least 1bp overlap
Analyse each of these novel “stranded” superloci separately.
Create subloci, ie group transcripts so to minimize the probability of mistakenly merging multiple gene loci due to chimeras. These groups are defined as follows:
- if the transcripts are multiexonic, they must share at least one intron, inclusive of the borders
- if the transcripts are monoexonic, they must overlap by at least 1bp.
- Monoexonic and multiexonic transcripts cannot be part of the same sublocus.
Select the best transcript inside each sublocus:
- Score the transcripts (see the section on scoring)
- Select as winner the transcript with the highest score and assign it to a monosublocus
- Discard any transcript which is overlapping with it, according to the definitions in the point above
- Repeat the procedure from point 2 until no transcript remains in the sublocus
Monosubloci are gathered together into monosubloci holders, ie the seeds for the gene loci. Monosubloci holders have more lenient parameters to group transcripts, as the first phase should have already discarded most chimeras. Once a holder is created by a single monosublocus, any subsequent candidate monosublocus will be integrated only if the following conditions are satisfied:
if the candidate is monoexonic, its exon must overlap at least one exon of a transcript already present in the holder
if the candidate is multiexonic and the holder contains only monoexonic transcripts, apply the same criterion, ie check whether its exons overlap the exons of at least one of the transcripts already present
if the candidate is multiexonic and the holder contains multiexonic transcripts, check whether one of the following conditions is satisfied:
- at least one intron of the candidate overlaps with an intron of a transcript in the holder
- at least one intron of the candidate is completely contained within an exon of a transcript in the holder
- at least one intron of a transcript in the holder is completely contained within an exon of a transcript in the holder.
- the cDNA overlap and CDS overlap between the candidate and the transcript in the holder are over a specified threshold.
Optionally, it is possible to tell Mikado to use a simpler algorithm, and integrate together all transcripts that share exon space. Such a simpler algorithm risks, however, chaining together multiple loci - especially in small, compact genomes.
Once the holders are created, apply the same scoring and selection procedure of the sublocus selection step. The winning transcripts are assigned to the final loci. These are called the primary transcripts of the loci.
Once the loci are created, track back to the original transcripts of the superlocus:
discard any transcript overlapping more than one locus, as these are probably chimeras.
For those transcripts that are overlapping to a single locus, verify that they are valid alternative splicing events using the class code of the comparison against the primary transcript. Transcripts are re-scored dynamically when they are re-added in this fashion, to ensure their quality when compared with the primary transcript.
- For coding loci, transcripts will be added as alternative splicing events only if they are in the same frame as the primary transcript. New in version 1.5.
If there are transcripts that do not overlap any of the final loci, create a new superlocus with the missed transcripts and perform the scoring and selection again on them, until no transcript is unaccounted for.
After the alternative splicing events have been defined, Mikado can optionally “pad” them. See the padding section for details.
Finally detect and either tag or discard fragments inside the initial superlocus (irrespective of strand):
- Check whether the primary transcript of any locus meets the criteria to be defined as a fragment (by default, maximum ORF of 30AA and maximum 2 exons - any transcript exceeding either criterion will be considered as non-fragment by default)
- If so, verify whether they are near enough any valid locus to be considered as a fragment (in general, class codes which constitute the “Intronic”, “Fragmentary” and “No overlap” categories).
- If these conditions are met, tag the locus as a fragment. If requested, Mikado will just discard these transcripts (advised).
These steps help Mikado identify and solve fusions, detect correctly the gene loci, and define valid alternative splicing events.
8.2. Definition of retained introns¶
When gathering transcripts into loci, Mikado will try to identify and tag transcripts that contain retained intron events. For our purposes, a retained intron event is an exon which:
- is part of a coding transcript but is not completely coding itself.
- if it is an internal exon, it completely spans the putative retained intron.
- if it is a terminal exon, it must start within the exon of the putative retained intron, and terminate within the intron.
- if it constitutes a monoexonic transcript, at least one of the two ends must reside within the bordering exons.
In addition to this, a transcript might be tagged as having its CDS disrupted by the retained intron event if:
- the non-coding part of the exon is in the 3’UTR and it begins within the intron
- the exon is 3’ terminal, coding and it ends within the intron.
Warning
The definition of a retained intron is stricty context dependent, i.e. the same exon will be regarded as a “retained intron” if the transcript is gathered together with other transcripts, but as non-retained if it were in isolation. It is therefore normal and expected that the associated metrics and scores will change, for a given transcript, across the various clustering stages.
8.3. Identification and breaking of chimeric transcripts¶
When a transcript contains more than one ORF, Mikado will try to determine whether this is due to a retained intron event or a frameshift (in which case the two ORFs are presumed to be mangled forms of an original, correct ORF for a single protein) or whether instead this is due to the fragment being polycystronic (in a prokaryote) or chimeric (in a eukaryote). The latter case is relatively common due to technical artefacts during sequencing and assembling of RNASeq reads.
A chimeric transcript is defined by Mikado as a model with multiple ORFs, where:
- all the ORFs share the same strand
- all the ORFs are non-overlapping.
In these situations, Mikado can try to deal with the chimeras in five different ways, in decreasingly conservative fashion:
- nosplit: leave the transcript unchanged. The presence of multiple ORFs will affect the scoring.
- stringent: leave the transcript unchanged, unless the two ORFs both have hits in the protein database and none of the hits is in common.
- lenient: leave the transcript unchanged, unless either the two ORFs both have hits in the protein database, none of which is in common, or both have no hits in the protein database.
- permissive: presume the transcript is a chimera, and split it, unless two ORFs share a hit in the protein database.
- split: presume that every transcript with more than one ORF is incorrect, and split them.
If any BLAST hit spans the two ORFs, then the model will be considered as a non-chimera because there is evidence that the transcript constitutes a single unit. The only case when this information will be disregarded is during the execution of the split mode.
These modes can be controlled directly from the pick command line, or during the initial configuration stage.
8.4. Transcript measurements and scoring¶
In order to determine the best transcript for each locus, Mikado measures each available candidate according to various different metrics and assigning a specific score for each of those. Similarly to RAMPART [Rampart], Mikado will assign a score to each transcript for each metric by assessing it relatively to the other transcripts in the locus. The particular feature rescaling equation used for a given metric depends on the type of feature it represents:
- metrics where higher values represent better transcript assemblies (“maximum”).
- metrics where lower values represent better transcript assemblies (“minimum”)
- metrics where values closer to a defined value represent better assemblies (“target”)
To allow for this tripartite scoring system with disparate input values, we have to employ rescaling equations so that each metric for each transcript will be assigned a score between 0 and 1. Optionally, each metric might be assigned a greater weight so that its maximum possible value will be greater or smaller than 1. Formally, let metric \(m\) be one of the available metrics \(M\), \(t\) a transcript in locus \(L\), \(w_{m}\) the weight assigned to metric \(m\), and \(r_{mt}\) the raw value of metric \(m\) for \(t\). Then, the score to metric \(m\) for transcript \(t\), \(s_{mt}\), will be derived using one of the following three different rescaling equations:
- If higher values are best:
- \(s_{mt} = w_{m} * (\frac{r_{mt} - min(r_m)}{max(r_m)-min(r_m)})\)
- If lower values are best:
- \(s_{mt} = w_{m} * (1 - \frac{r_{mt} - min(r_m)}{max(r_m)-min(r_m)})\)
- If values closer to a target \(v_{m}\) are best:
- \(s_{mt} = w_{m} * (1 - \frac{|r_{mt} - v_{m}|}{max(|r_{m} - v_{m}|)})\)
- Finally, the scores for each metric will be summed up to produce a final score for the transcript:
- \(s_{t} = \sum_{m \forall m \in M} s_{mt}\).
Not all the available metrics will be necessarily used for scoring; the choice of which to employ and how to score and weight each of them is left to the experimenter, although Mikado provides some pre-configured scoring files. Values that are guaranteed to be between 0 and 1 (e.g. a percentage value) can be used directly as scores, by setting the use_raw parameter as true for them.
Details on the structure of scoring files can be found in a dedicated section; we also provide a tutorial on how to create your own scoring file.
Important
The scoring algorithm is dependent on the other transcripts in the locus, so each score should not be taken as an absolute measure of the reliability of a transcript, but rather as a measure of its relative goodness compared with the alternatives. Shifting a transcript from one locus to another can have dramatic effects on the scoring of a transcript, even while most or all of the underlying metric values remain unchanged. This is why the score assigned to each transcript changes throughout the Mikado run, as transcripts are moved to subloci, monoloci and finally loci.
8.5. Padding transcripts¶
Mikado has the ability of padding transcripts in a locus, so to uniform their starts and stops, and to infer the presence of missing exons from neighbouring data. The procedure is similar to the one employed by PASA and functions as follows:
- A transcript can function as template for a candidate if:
- the candidate’s terminal exon falls within an exon of the template
- the extension would enlarge the candidate by at most “ts_distance” basepairs (not including introns), default 2000 bps
- the extension would add at most “ts_max_splices” splice sites to the candidate, default 2.
- A graph of possible extensions is built for both the 5’ end and the 3’ end of the locus. Transcripts are then divided in extension groups, starting with the outmost (ie the potential template for the group). Links that would cause chains (e.g. A can act as template for B and B can act as template for C, but A cannot act as template for C) are broken.
- Create a copy of the transcripts in the locus, for backtracking.
- Start expanding each transcript:
- Create a copy of the transcript for backtracking
- Calculate whether the 5’ terminal exon should be enlarged:
- if the transcript exon terminally overlaps a template exon, enlarge it until the end of the template
- If the template transcript has multiple exons upstream of the expanded exon, add those to the transcript.
- Calculate the number of bases that have been added upstream to the cDNA of the transcript
- Calculate whether the 3’ terminal exon should be enlarged:
- if the transcript exon terminally overlaps a template exon, enlarge it until the end of the template
- If the template transcript has multiple exons downstream of the expanded exon, add those to the transcript.
- Calculate the number of bases that have been added downstream to the cDNA of the transcript
- If the transcript is coding:
- Calculate the new putative CDS positions in the transcript, using the memoized amount of added basepairs downstream and upstream
- Calculate the new CDS, keeping the same frame as the original transcript. If the transcript is incomplete, this might lead to find the proper start and stop codons
- If we find an in-frame stop codon, the expansion would lead to an invalid transcript. Backtrack.
- Recalculate metrics and scores.
- Check whether we have made any transcript an invalid alternative splicing event; possible common causes include:
- Having created a retained intron
- Having expanded the number or size of the UTR so that the transcripts are no longer viable
- If any of the non-viable transcripts is either the primary transcript or one of the templates, remove the current templates from the locus and restart the analysis.
- Discard all the non-viable transcripts that are neither the primary nor templates.
When calculating the new ORF, Mikado will use the same codon table selected for the serialisation step.
This option is normally activated, with the parameters:
- Default maximum splice sites that can be crossed: 2
- Default maximum basepair distance: 1000
Note
please consider that the parameters above refer to the expansion on both sides of the transcript. So the parameters above allow transcripts to be expanded by up to 2000 bps, ie 1000 in both directions.
This option has been written for using Mikado in conjunction with ab initio predictions, but it can be used fruitfully also with transcript assemblies.
Warning
Please note that some of the metrics might become invalid after the padding. In particular, BLASTX results will be invalid as the query sequence will have changed.
The options related to padding can be found under the pick section in the configuration file.
8.6. Technical details¶
Most of the selection (ie “pick”) stage of the pipeline relies on the implementation of the objects in the loci submodule. In particular, the library defines an abstract class, “Abstractlocus”, which requires all its children to implement a version of the “is_intersecting” method. Each implementation of the method is specific to the stage. So the superlocus class will require in the “is_intersecting” method only overlap between the transcripts, optionally with a flanking and optionally restricting the groups to transcripts that share the same strand. The sublocus class will implement a different algorithm, and so on. The scoring is effectuated by first asking to recalculate the metrics (.calculate_metrics) and subsequently to calculate the scores (.calculate_scores). Mikado will try to cache and avoid recalculation of metrics and scores as much as possible, to make the program faster.
Metrics are an extension of the property construct in Python3. Compared to normal properties, they are distinguished only by three optional descriptive attributes: category, usable_raw, and rtype. The main reason to subclass property is to allow Mikado to be self-aware of which properties will be used for scoring transcripts, and which will not. So, for example, in the following snippet from the Transcript class definition:
@property
def combined_cds(self):
"""This is a list which contains all the non-overlapping CDS
segments inside the cDNA. The list comprises the segments
as duples (start,end)."""
return self.__combined_cds
@combined_cds.setter
def combined_cds(self, combined):
"""
Setter for combined_cds. It performs some basic checks,
e.g. that all the members of the list are integer duplexes.
:param combined: list
:type combined: list[(int,int)]
"""
if ((not isinstance(combined, list)) or
any(self.__wrong_combined_entry(comb) for comb in combined)):
raise TypeError("Invalid value for combined CDS: {0}".format(combined))
@Metric
def combined_cds_length(self):
"""This property return the length of the CDS part of the transcript."""
c_length = sum([c[1] - c[0] + 1 for c in self.combined_cds])
if len(self.combined_cds) > 0:
assert c_length > 0
return c_length
combined_cds_length.category = "CDS"
@Metric
def combined_cds_num(self):
"""This property returns the number of non-overlapping CDS segments
in the transcript."""
return len(self.combined_cds)
combined_cds_num.category = "CDS"
@Metric
def has_start_codon(self):
"""Boolean. True if the selected ORF has a start codon.
:rtype: bool"""
return self.__has_start_codon
@has_start_codon.setter
def has_start_codon(self, value):
"""Setter. Checks that the argument is boolean.
:param value: boolean flag
:type value: bool
"""
if value not in (None, False, True):
raise TypeError(
"Invalid value for has_start_codon: {0}".format(type(value)))
self.__has_start_codon = value
has_start_codon.category = "CDS"
Mikado will recognize that “derived_children” is a normal property, while “combined_cds_length”, “combined_cds_num” and “has_start_codon” are Metrics (and as such, we assign them a “category” - by default, that attribute will be None.). Please note that Metrics behave and are coded like normal properties in any other regard - including docstrings and setters/deleters.
The requirements expression is evaluated using eval.
Warning
While we took pains to ensure that the expression is properly sanitised and inspected before eval, Mikado might prove itself to be permeable to clever code injection attacks. Do not execute Mikado with super user privileges if you do not want to risk from such attacks, and always inspect third-party YAML scoring files before execution!