1. Introduction¶
Numerous algorithms have been proposed to analyse RNA-Seq data, both in terms of aligning the reads to a reference genome ([TopHat2], [STAR], [Hisat]) or to assemble them to infer the sequence and structure of the original molecules present in the sample. The latter phase can be performed either by using alignment data against the reference genome ([Cufflinks], [StringTie], [Class2], [Trinity]) or in the absence of such information ([Trinity], [Oases], [Bridger], ). Each of these methods has to contend with numerous sources of variability in the RNA-Seq data:
- alternative splicing events at the same locus
- extremely variable expression and therefore sequencing depth at different loci
- presence of orthologous genes with very similar cDNAs.
- tandem duplicated genes which can be artefactually reconstructed as a single, fused entity
Multiple assessments of these methods on real and simulated data. In particular, the RGASP consortium promoted a competition among some of the most popular methods in 2012 [RGASP]; the results showed that the accuracy of the methods was dependent on the input data, the expression levels of the genes, and on the species under analysis. A different concern regards the completeness of each of the assembly methods, ie whether methods that are not necessarily the best across all gene loci might nonetheless be capable of outperforming other competitors for specific contigs. Recently, approaches have been proposed to integrate multiple RNA-Seq assemblies by inferring their protein-coding content and collapsing them accordingly [EviGeneTobacco] or to determine the best proposed assembly using multiple measures related to how the transcript compares to the input RNA-Seq reads ([Transrate], [RSEMeval]).
A different but related line of research has focused on how to integrate data coming from multiple samples. While some researchers advocate for merging together the input data and assembling it [Trinity], others have developed methods to integrate multiple assemblies into a single coherent annotation ([CuffMerge], Stringtie-merge). Another common approach relies on a third party tool to integrate multiple assemblies together with other data - typically protein alignments and ab initio predictions - to select for the best model. The latter approach has been chosen by some of the most popular pipelines for genome annotation in the last years ([EVM], [Maker]).
Our tool, Mikado, contributes to this area of research by proposing a novel algorithm to integrate multiple transcript assemblies, leveraging additional data regarding the position of ORFs (generally derived with Transdecoder [Trinity]), sequence similarity (derived with BLAST [Blastplus]) and reliable splicing junctions (generally derived using Portcullis [Portcullis]). Through the combination of these input data sources, we are capable of identifying artefacts - especially gene fusions and small fragments - and retrieve the original transcripts in each locus with high accuracy and sensitivity.
An important caveat is that our approach does not look for the best candidate in terms of the input data, as Transrate [Transrate] or RSEM-Eval [RSEMeval] do. Rather, our approach is more similar to EvidentialGene [EviGeneTobacco] as Mikado will try to find the candidates most likely to represent the isoforms which would be annotated as canonical by a human annotator. Biological events such as intron retentions or trans-splicing that might be present in the sample are explicitly selected against, in order to provide a clean annotation of the genome.
1.1. Overview¶
Mikado analyses an ensemble of RNA-Seq assemblies by looking for overlapping transcripts based on their genomic position. Such clusters of genes, or superloci, are then analysed to locate the gene loci that will form the final annotation. In order, the steps of the pipeline are as follows:
- In the first step,
mikado preparewill combine assemblies from multiple sources into a single coherent, filtered dataset. - In the second step,
mikado serialisewill collect data from multiple sources (Portcullis, Transdecoder, BLASTX) and load it into a database for fast integration with the input data. - In the final step,
mikado pickwill integrate the prepared transcripts with the serialised additional data to choose the best transcripts.
By far, the most important source of data for Mikado is the ORF calling, which can be executed with Prodigal or Transdecoder. This is because many of the algorithms for finding and splitting chimeras depend on the detection of the ORFs on the transcript sequences. Moreover, the ORF features are a very important part of the scoring mechanism. Please refer to the Algorithms section for more details on how Mikado operates.
Using these multiple data sources, and its distinctive iterative method to identify and disentangle gene loci, Mikado is capable of bringing together methods with very different results, and interpret the composition of a locus similarly to how a manual annotator would. This leads to cleaned RNA-Seq annotations that can be used as the basis of the downstream analysis of choice, such as eg a hint-based ab initio annotation with Augustus [Augustus] or Maker2 [Maker].
Mikado in action: recovering fused gene loci
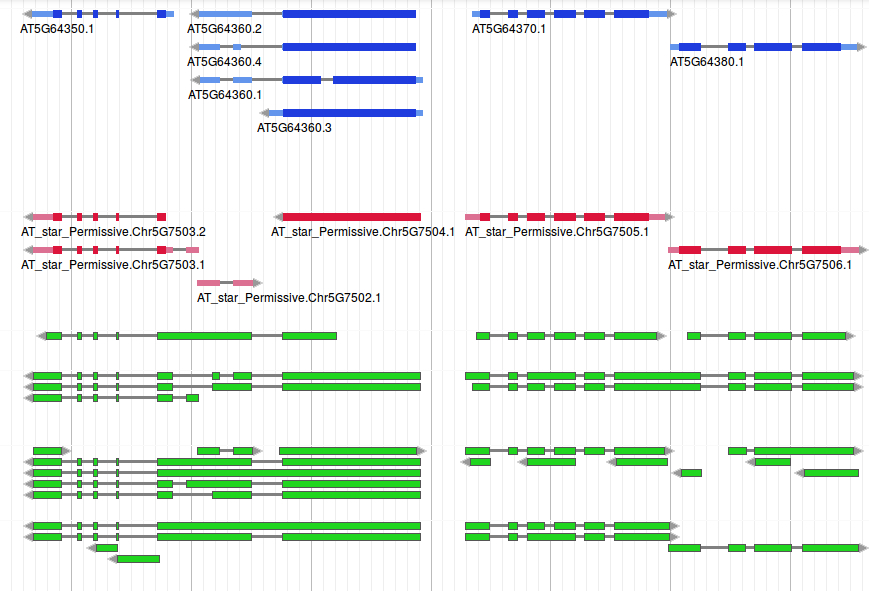
Using data from the ORFs, Mikado (in red) is capable of identifying and breaking artefactual gene fusions found by the assemblers (in green) in this locus. This allows to report the correct gene loci, even when no method was capable of retrieving all the loci in isolation, and some of the correct genes were not present at all. Coding sections of Mikado models are in dark red, UTR segments are in pale red. The reference annotation is coloured in blue, with the same colour scheme - dark for coding regions, pale for UTRs.
1.2. The command-line interface¶
The Mikado suite provides two commands: mikado and daijin. The former provides access to the main functionalities of the suite:
$ mikado --help
usage: Mikado [-h] {configure,prepare,serialise,pick,compare,util} ...
Mikado is a program to analyse RNA-Seq data and determine the best transcript
for each locus in accordance to user-specified criteria.
optional arguments:
-h, --help show this help message and exit
Components:
{configure,prepare,serialise,pick,compare,util}
These are the various components of Mikado:
configure This utility guides the user through the process of
creating a configuration file for Mikado.
prepare Mikado prepare analyses an input GTF file and prepares
it for the picking analysis by sorting its transcripts
and performing some simple consistency checks.
serialise Mikado serialise creates the database used by the pick
program. It handles Junction and ORF BED12 files as
well as BLAST XML results.
pick Mikado pick analyses a sorted GTF/GFF files in order
to identify its loci and choose the best transcripts
according to user-specified criteria. It is dependent
on files produced by the "prepare" and "serialise"
components.
compare Mikado compare produces a detailed comparison of
reference and prediction files. It has been directly
inspired by Cufflinks's cuffcompare and ParsEval.
util Miscellaneous utilities
Each of these subcommands is explained in detail in the Usage section.
daijin instead provides the interface to the Daijin pipeline manager, which manages the task of going from a dataset of multiple reads to the Mikado final picking. This is its interface:
$ daijin --help
usage: A Directed Acyclic pipeline for gene model reconstruction from RNA seq data.
Basically, a pipeline for driving Mikado. It will first align RNAseq reads against
a genome using multiple tools, then creates transcript assemblies using multiple tools,
and find junctions in the alignments using Portcullis.
This input is then passed into Mikado.
[-h] {configure,assemble,mikado} ...
optional arguments:
-h, --help show this help message and exit
Pipelines:
{configure,assemble,mikado}
These are the pipelines that can be executed via
daijin.
configure Creates the configuration files for Daijin execution.
assemble A pipeline that generates a variety of transcript
assemblies using various aligners and assemblers, as
well a producing a configuration file suitable for
driving Mikado.
mikado Using a supplied configuration file that describes all
input assemblies to use, it runs the Mikado pipeline,
including prepare, BLAST, transdecoder, serialise and
pick.